Studying fundamental aspects of gene regulation and molecular network organization: systematic identification of regulatory elements in physiological conditions and development of dedicated bioinformatics approaches and tools
|
This research axis focus on :
|
Deciphering the functional landscape of the coding genome
RNA BIndiNG PrOtein - RNA network
|
Novel high throughput methods recently led to the discovery of an unprecedented number of proteins able to bind RNAs and reciprocally, thereby revealing a terra incognita of possible regulatory roles. By coupling predictions of RNA-protein interactions and statistical analyses, we recently showed that the scaffolding of protein complex is a prevalent function of long non-coding RNAs (collaboration with G.G. Tartaglia, CRG, Barcelona, Spain and I. Bozzoni, La Sapienza, Rome, Italy). |
Protein-protein interaction (PPI) variant networks
|
|
 Genetic variations in coding sequences of the genome can lead to cascades of molecular and cellular consequences ultimately influencing the phenotype. Non-synonymous SNPs can functionally affect proteins by impairing their integrity, stability and molecular function. As proteins seldom act alone but interact with other macromolecules to perform their cellular functions, these genetic variations can modify protein interactions, thereby rewiring the PPI network in the cell. What is the influence of genetic variation on PPI networks? How does it affect protein interactions, biological processes and in turn, phenotypes? To answer these questions, we model the interactomes of inbred Drosophila melanogaster lines in a population-wide analysis of interactomes.
Genetic variations in coding sequences of the genome can lead to cascades of molecular and cellular consequences ultimately influencing the phenotype. Non-synonymous SNPs can functionally affect proteins by impairing their integrity, stability and molecular function. As proteins seldom act alone but interact with other macromolecules to perform their cellular functions, these genetic variations can modify protein interactions, thereby rewiring the PPI network in the cell. What is the influence of genetic variation on PPI networks? How does it affect protein interactions, biological processes and in turn, phenotypes? To answer these questions, we model the interactomes of inbred Drosophila melanogaster lines in a population-wide analysis of interactomes.Deciphering the functional organization of the genome
Enhancers /silencers
|
|
 While hundreds of genomes have been sequenced and annotated, while multiple layers of various sequencing assays have revealed the extent of the “non-coding” regions, little is known about the exact regulatory roles of those regions in gene regulation. We will continue to further characterise the regulatory landscape across three species (ReMap), human, mouse and drosophila, all used in the research undertaken at TAGC. The challenges for the next decade will be to decipher and explain how this regulatory landscape models our genome. Specifically, by computational and genomics approaches, we plan on dissecting further enhancers and silencers functions as well as their roles in the complexity of our genome. In particular, by taking advantage of our recently developed high-throughput reporter assay (CapStarr-seq), we will assess enhancer activity in different cellular contexts, including cell stimulation, transcription factor perturbation and pathological models (see also Axis 2). We will also repurpose this technique in order to identify and characterise silencer elements.
While hundreds of genomes have been sequenced and annotated, while multiple layers of various sequencing assays have revealed the extent of the “non-coding” regions, little is known about the exact regulatory roles of those regions in gene regulation. We will continue to further characterise the regulatory landscape across three species (ReMap), human, mouse and drosophila, all used in the research undertaken at TAGC. The challenges for the next decade will be to decipher and explain how this regulatory landscape models our genome. Specifically, by computational and genomics approaches, we plan on dissecting further enhancers and silencers functions as well as their roles in the complexity of our genome. In particular, by taking advantage of our recently developed high-throughput reporter assay (CapStarr-seq), we will assess enhancer activity in different cellular contexts, including cell stimulation, transcription factor perturbation and pathological models (see also Axis 2). We will also repurpose this technique in order to identify and characterise silencer elements.Regulatory variants
|
Our better understanding of regulatory region and large amount of available data allow us to systematically study the influence of regulatory variation on complex phenotypes. Based on the strengths of the laboratory (Genome-wide association studies, analysis of regulatory regions), we are working on new bioinformatics and experimental methods to understand and predict regulatory variants. On the one hand, we are integrating phenotypic variant information with chromatin and sequence data to gain insights into the molecular properties of functional regulatory variants. We use this knowledge to develop predictive in silico models. On the other hand, we are implementing high-throughput reporter assays in order to test thousands of regulatory variants in parallel. Complex diseases are largely influenced by genomic variants. Therefore, a number of complex diseases are studied in the thematics Axis 2 from the point of view of functional variants (Malaria, Sepsis). The bioinformatics and experimental tools developed here will be thus be shared and tested in collaboration with these laboratory members. |
Chromatin organization and replication
|
We have previously shown that the replication origins can be grouped into different classes with distinct organization, chromatin environment, and sequence motifs. The current challenge is to specifically dissect the replications origins located in regulatory regions (enhancers, promoters) and address how those initiation sites are replicated with regards to gene expression,regulation and cell differentiation (in collaboration with M. Mechali, IGH, Montpellier). We also pursue our collaboration with S. Khochbin and S. Rousseaux (Institut Albert Bonniot, Grenoble, France) on the molecular basis of male germ cells genome programming, to describe precisely the chromatin remodeling event taking place through spermiogenesis (funded by FRM). Our mid-term objective is to integrate our in-house data with public data encompassing MNase-Seq, RNA-Seq, whole-genome bisulfite sequencing (WGBS), ChIP-Seq from various histone marks and bromo-domain proteins to define precisely the molecular bases of male germ cell genome packaging. |
Deciphering the functional role of non-coding transcripts (eRNA, LncRNA)
enhancer RNA-RBP
|
In the light of recent findings (ReMap catalogue), we will address the functional role of a specific group of enhancers, the transcribed enhancers. Since some ‘active’ enhancers are transcribed bidirectionally and produce short capped RNA called eRNA, we will investigate their functional role notably by analyzing their propensity to bind RNA Binding Protein (RBPs). |
Long non-coding RNA
|
Based on in-house RNA-seq data obtained from normal thymocytes and leukemic cells, we will identify lncRNAs dysregulated in leukemia and perform functional assays to study their potential implications in oncogenesis (in collaboration with V. Asnafi, Necker Hospital, Paris, France; funded by ITMO-Plan Cancer). We will also prioritise potentially relevant lncRNAs based on epigenomics profiles we have obtained within the Blueprint consortium. In addition, the prevalence of the role of lncRNAs as scaffolding molecule of protein complexes has been investigated using an original bioinformatic approach coupling predictions of RNA-protein interactions (in collaboration with G.G. Tartaglia, CRG, Barcelona, Spain) and protein-protein interaction network analyses (funded by A*MIDEX grant). |
Deciphering genetic networks in a model organism
|
|
 We develop systems genetics approaches of cardiac function and aging in flies leveraging on the identification of natural variants in the DGRP population of sequenced inbred lines (Drosophila Genetic Reference Panel; dgrp.gnets.ncsu.edu). This resource makes it possible to carry out association analyzes for quantitative traits while being able to identify precisely the variants (SNPs) involved. The DGRP lines are tested for their cardiac phenotypes as a function of age and the variants (SNPs) associated with a phenotype of increased or decreased sensitivity of the heart to cardiac aging is carried out by a genome-wide association analysis (GWAS). We seek for epistasis relationships among variants in different contexts and develop a method for the identification of rare non-synonymous variants that are not considered in GWAS approaches. This genetic data are integrated with transcriptomic data, PPI networks and regulatory networks to identify novel genes affecting cardiac function and aging. This work is performed in collaboration with Rolf Bodmer’s lab (Stanford Burnham Medical Research Institute, La Jolla, California, USA).
We develop systems genetics approaches of cardiac function and aging in flies leveraging on the identification of natural variants in the DGRP population of sequenced inbred lines (Drosophila Genetic Reference Panel; dgrp.gnets.ncsu.edu). This resource makes it possible to carry out association analyzes for quantitative traits while being able to identify precisely the variants (SNPs) involved. The DGRP lines are tested for their cardiac phenotypes as a function of age and the variants (SNPs) associated with a phenotype of increased or decreased sensitivity of the heart to cardiac aging is carried out by a genome-wide association analysis (GWAS). We seek for epistasis relationships among variants in different contexts and develop a method for the identification of rare non-synonymous variants that are not considered in GWAS approaches. This genetic data are integrated with transcriptomic data, PPI networks and regulatory networks to identify novel genes affecting cardiac function and aging. This work is performed in collaboration with Rolf Bodmer’s lab (Stanford Burnham Medical Research Institute, La Jolla, California, USA).Databases
REMAP
|
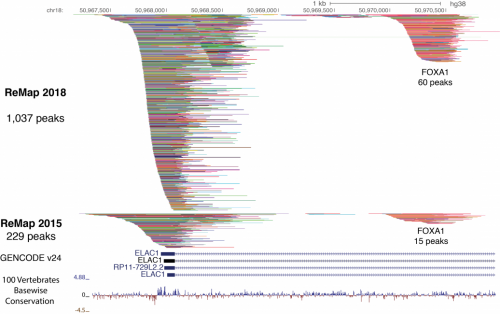
ReMap (remap.cisreg.eu) is an integrative analysis of transcriptional regulators ChIP-seq experiments from both “Public” (GEO / ArrayExpress) and Encode datasets. The ReMap atlas consists of 80 million peaks from 485 transcription factors (TFs), transcription coactivators (TCAs) and chromatin-remodeling factors (CRFs). The ReMap atlas is available to browse or download either for a given TF or cell line, or for the entire dataset.
|
MoonDB
|
MoonDB is a database of Extreme Multifunctional proteins (EMFs) that we recently identified as proteins involved in very dissimilar functions from PPI networks. In addition to functional informations on the 430 identified EMFs, a dataset of 38 literature curated proteins is provided. |









